

Resolving the Challenges of Failure Mode and Effects
Analysis (FMEA) for Medical Devices
by Alec Alpert

⇓ Download this article as PDF
Without a doubt, medical device manufacturers face multiple challenges. Stringent regulations, intense competition, and accelerated product development cycles are just a few of the issues manufacturers must handle. However, effective risk management throughout the product's entire life cycle is undoubtedly paramount.
Risk management helps decision-makers understand risk while directing their focus on risk tolerance. Although comparing technical and economic practicability against risks and benefits helps to control the residual risk, it is important to note that risk can never be completely eliminated.
All medical device manufactures are required to have a robust risk analysis system within the Quality System, as stipulated by these key regulations and standards:
- 21 CFR 820: Quality System Regulation
- ISO 13485: Medical Devices: Quality Management Systems Requirements
- ISO 14971: Application of Risk Management to Medical Devices
- ISO 9001: Quality Management Systems Requirements
- IEC 60601: General Requirements for Basic Safety and Essential Performance
- IEC 62304: Medical Device Software: Software Life-Cycle Processes
- EU MDR 2017/745: Regulation of the European Parliament and of the Council
To reduce risk, it is essential to identify hazards, evaluate the probability of potential consequences, and then estimate the risk. The following three tools are recommended by the FDA and widely used by medical device manufacturers:
- Failure Mode and Effects Analysis (FMEA)
- Fault Tree Analysis (FTA)
- Hazard and Operability Analysis (HAZOP)
This article focuses on the FMEA’s bottom-up approach. There are several varieties:
- Design FMEA
- Use FMEA
- Process FMEA
- Service FMEA
- Application FMEA
- Hardware FMEA
- Software FMEA
These diverse FMEAs focus on different aspects of medical devices. In this article, we will explore the first three: Design FMEA, Use FMEA, and Process FMEA.
- Design FMEA (DFMEA): This method identifies, prioritizes, and mitigates the device design and assembly failure modes.
- Use FMEA (UFMEA): This method identifies, prioritizes, and mitigates the product use and functional failure modes. A use failure mode occurs when the design fails to perform as intended due to incorrect use by the consumer. Incorrect use can occur when the user fails to follow the guidelines provided in the Instructions for Use (IFU).
- Process FMEA (PFMEA): This method identifies, prioritizes, and mitigates the process and equipment failure modes.
Design FMEA Concept
Design FMEA analyzes all possible failure modes for each design component, and it identifies how the component or sub-system can fail to perform its function(s) and affect the end user. Design FMEA is a bottom-up analysis of possible failure modes, and it addresses concerns like the ones below.
- How can this component or assembly fail?
- What is the effect on the end user in terms of potential harm?
- What are the possible causes of this failure?
- What is the anticipated percentage of patients who may be harmed by this failure?
- What actions can be taken to prevent or mitigate this failure mode?
Use FMEA Concept
Use FMEA analyzes possible failure modes that may occur by moving step-by-step through the use of the product. Use FMEA identifies those failures and their effects from the end user’s viewpoint. The Use FMEA is a top-down analysis of possible failure modes, and it addresses concerns like the ones below.
- How can the product fail when it is in use?
- What is the effect on the end user in terms of potential harm?
- What are the causes of this failure, including known misuses?
- What is the anticipated percentage of patients who may be harmed by this failure?
- What actions can be taken to prevent or mitigate this failure mode?
The Use FMEA usually includes a clinician review to ensure the correct assessment of possible failure effects, along with their associated severities.
Process FMEA Concept
Process FMEA analyzes the possible failure modes in each process, and it identifies how the process can affect the end user by failing to meet required specifications. Process FMEA is caused by failure modes (identified in the Use or Design FMEA) related to the design’s manufacturing processes. The Process FMEA is a bottom-up analysis of work instructions, equipment settings, material handling, and fixtures. It addresses concerns like the ones below:
- What portions of the process could be completed incorrectly?
- What process settings or fixtures introduce risk or unacceptable variation, therefore contributing to potential failure?
- In what ways can a part be out of specification in each stage of operation?
- What are the effects of these possible risks on the process and product in terms of failure or design risk?
- What is the percentage of patients who may be harmed by this failure?
- What actions can be taken to prevent or mitigate identified failure modes?
Performing FMEAs
Performing FMEAs is an elaborate process requiring good preparation and collaboration within the FMEA team. Depending on the complexity of the product, many brainstorming sessions may be needed to complete the FMEA. These sessions have the potential to be tedious. A good rule of thumb is to have multiple one-hour sessions, as sessions exceeding an hour can tire the participants and reduce the brainstorming efficiency.
The FMEA team must include subject matter experts who possess thorough knowledge of the product and its use.
It is important to note that all brainstorming sessions need to be regimented, and all team members or their designees must be formally invited to confirm attendance. After each meeting, the facilitator should publish detailed meeting minutes and file them in the Risk Management File, as appropriate.
Excel spreadsheets are popular tools for recording FMEA details during brainstorming sessions. This technology facilitates real-time updates, and the user’s screen can be projected onto a larger screen via projectors or dual-monitor connections.
Below are the typical steps in the FMEA process.
The project management team appoints the FMEA facilitator.
- The facilitator forms the FMEA cross-functional team and schedules meetings.
- Depending on the project’s phase, the facilitator distributes applicable documents. These records may include the following documents:
- Customer requirements
- Assembly drawings
- Component drawings
- Engineering specifications
- Hardware specifications
- Software specifications
- Schematics
- Labeling explanations
- Instruction manuals
- Past FMEAs
- Published literature
The FMEA team then moves forward with the following steps:
- Possible failure modes are brainstormed.
- The effects of the failure modes with respect to the end user are analyzed.
- Possible causes of failure modes are explored and determined.
- The severity, occurrence, and detection ranking is determined for the Process FMEA.
- Risk classifications are determined.
- The results are analyzed, and necessary risk mitigation tasks are identified.
- Risk mitigation begins.
- After the risk mitigation is complete, the team should reevaluate its occurrence, detection, and severity.
Although each company may have different formats for the FMEA summary table, the essential items for the hardware DFMEA can be arranged like the below table.
DFMEA Summary Table
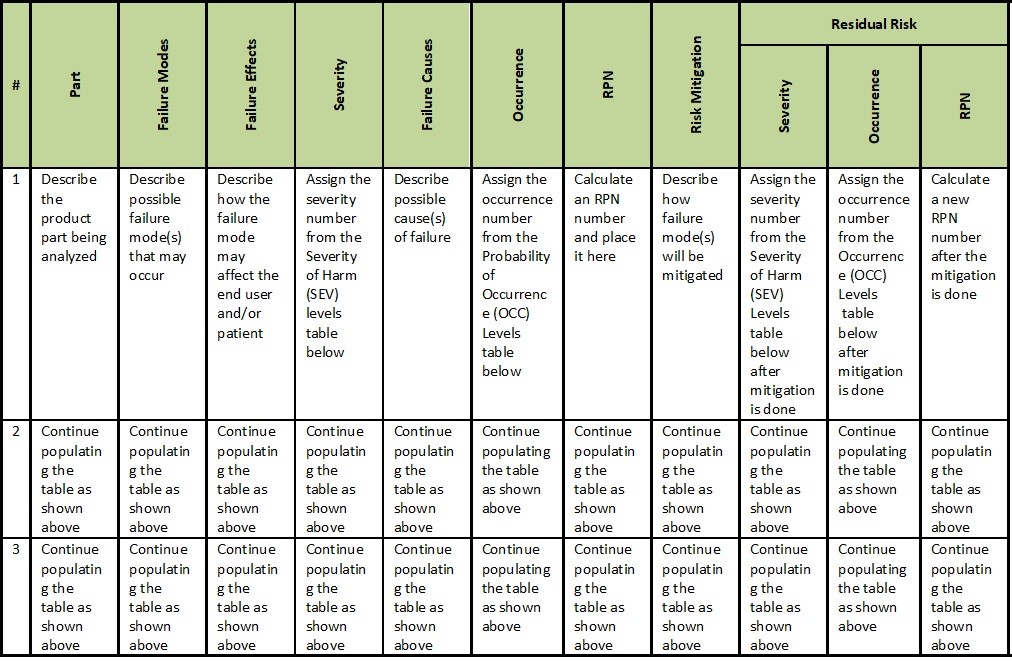
For the UFMEA, the above table should be modified to replace the “Part” column with “Clinical Step” or any other name that appropriately reflects your needs.
For the PFMEA, the table above should be modified by adding a “Detection” column.
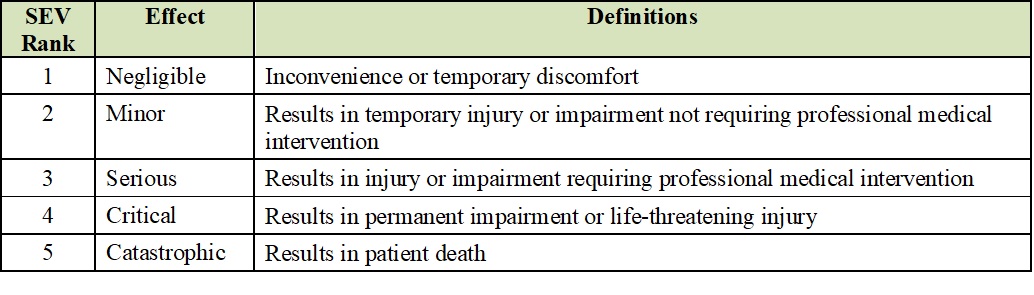
ISO 14971:2012 provides good guidance on risk qualitative and quantitative assessments, including “risk evaluation matrices” in section D.4, Risk Evaluation and Risk Acceptability. These matrices can be used as generic templates to create summary tables for the Severity (SEV) and Probability of Occurrence (OCC). Ranking columns can be added to calculate Risk Priority Numbers (RPN) numbers. Examples of these templates are depicted in the below tables.
Severity of Harm (SEV) Levels
Probability of Occurrence (OCC) Levels

Once SEV and OCC have been determined via the brainstorming sessions, RPNs can be calculated by multiplying SEV by OCC for each failure mode.
RPN = SEV x OCC
For PFMA, the formula will also include DET.
RPN = SEV x OCC x DET
The next step is to associate RPN numbers with the risk level. There may be several risk levels:
- Negligible
- Low
- Medium
- High
- Very High
Each company needs to define those risk levels and determine which are not acceptable, based on the RPN score. The unacceptable levels will require mitigation, along with a possible risk/benefit analysis.
Additionally, the risk levels and their associated RPN, SEV, OCC, and DET values can be entered in a concise Risk Evaluation Matrix. A basic template can be found in ISO 14971:2012, Figure D.5.
A customized Risk Evaluation Matrix from based on ISO 14971:2012 is below. It includes numerical values for the Severity and Probability of Occurrence levels and color coding to signify different risk levels.
Risk Evaluation Matrix
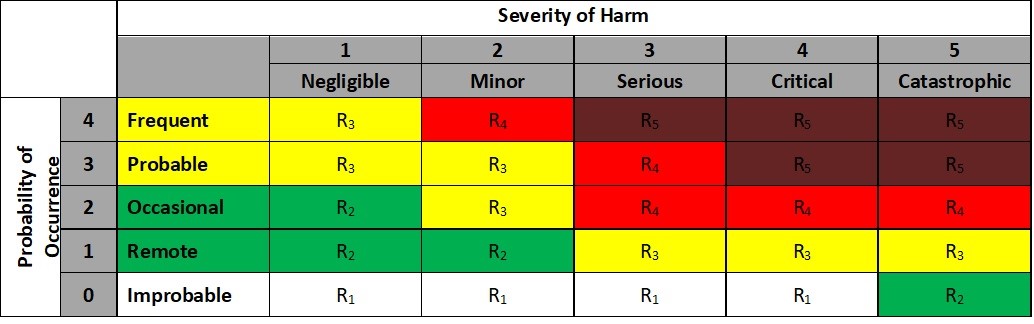
The R letters in the table represent different risk levels, which range from negligible (R1) to very high (R5). The company’s FMEA procedure has to spell out which risk levels are unacceptable. Levels that are unacceptable require risk reduction and a possible risk/benefit analysis.
Risk Mitigation
The EN ISO 14971:2012 standard states the following regarding risk reduction.
Risk reduction "as far as possible" versus "as low as reasonably practicable":
a) Annex D.8 to ISO 14971, referred to in 3.4, contains the concept of reducing risks "as low as
reasonably practicable" (ALARP concept). The ALARP concept contains an element of economic
consideration.
b) However, the first indent of Section 2 of Annex I to Directive 93/42/EEC and various particular
Essential Requirements require risks to be reduced "as far as possible" without there being room for
economic considerations.
c) Accordingly, manufacturers and Notified Bodies may not apply the ALARP concept with regard to
economic considerations.
According to EN ISO14971:2012, risk mitigation should seek to reduce risk “as far as possible.” Risk mitigation/reduction actions must be documented in the FMEA. If there are associated documents, all must be referenced in the FMEA.
Typical risk reduction actions are listed below.
- Design refinement
- Redesign
- Use of more reliable parts
- Software changes
- Design testing and statistical analysis
- Redesign of the manufacturing processes
- Process control
- Labeling
Residual Risk
Once risk mitigation actions are proposed and completed, the post-mitigation risk must be evaluated in the FMEA. These risks must be documented and reduced as low as reasonably practicable (ALARP), or as far as possible, depending on which version of ISO 14971 was referenced. Company’s policies and procedures should stipulate which ISO version is to be used.
After all risk control measures and mitigations have been implemented and verified, a risk/benefit analysis must be conducted and documented by the team. The purpose of this analysis is to determine if the medical benefits of the product outweigh the overall residual risk for specific risks that cannot be mitigated. The results of this review must be included in the Risk Management Report.
Sections D.6 and D.7. of ISO standard 14971:2012 provide guidelines on how to tackle risk/benefit analyses.
Further, the FDA published their own version of benefit-risk analysis in December 2016. This publication is called: Factors to Consider Regarding Benefit-Risk in Medical Device Product Availability, Compliance, and Enforcement - Decisions Guidance for Industry and Food and Drug Administration Staff. This is recommended reading for anyone involved with risk analysis, and it can be accessed here: https://bit.ly/2O7Si57.
The FMEA Life Cycle
Below are the typical FMEA life cycle milestones:
- The Design and Use FMEAs are to be initiated in the earliest stages of product development. However, these FMEAs must be approved prior to the beginning of the design verification process.
- The Process FMEA analysis must be approved prior to the initiation of process validation and commercialization.
- The FMEA analysis must be continuously updated by the team throughout the development of the device. These updates should be based on knowledge gained from the progress made in the device development, testing, and verification process.
- All FMEAs must revisited after product commercialization to ensure that field feedback is addressed.